How India’s Medical Device Regulations Are Evolving in 2025: What Manufacturers Must Know
Explore India’s 2025 med tech regulatory compliance changes, CDSCO updates, device classification, and steps to stay compliant in the new era.
Accorp Compliance Team
Our team of compliance experts specializes in PCI DSS, SOC 2, and other security frameworks to help businesses achieve and maintain compliance.
India’s MedTech sector is entering a decisive phase. With the market projected to surpass USD 50 billion in the next few years, the Central Drugs Standard Control Organization (CDSCO) and the Ministry of Health are tightening medical device regulations to ensure global-quality standards and patient safety. For manufacturers, importers, and distributors, 2025 brings both opportunities and responsibilities.
The Shift Toward a Regulated MedTech Ecosystem
For decades, India’s medical device sector operated with limited oversight. However, under the Medical Devices Rules (MDR) 2017 and subsequent amendments, more devices are now regulated as “drugs” under the Drugs and Cosmetics Act. This expansion ensures quality, safety, and post-market accountability across the supply chain.
As of 2025, over 600 devices fall under mandatory registration, and the CDSCO continues to integrate more product categories—particularly in diagnostics and digital health technology.
Key Updates in 2025 You Need to Know
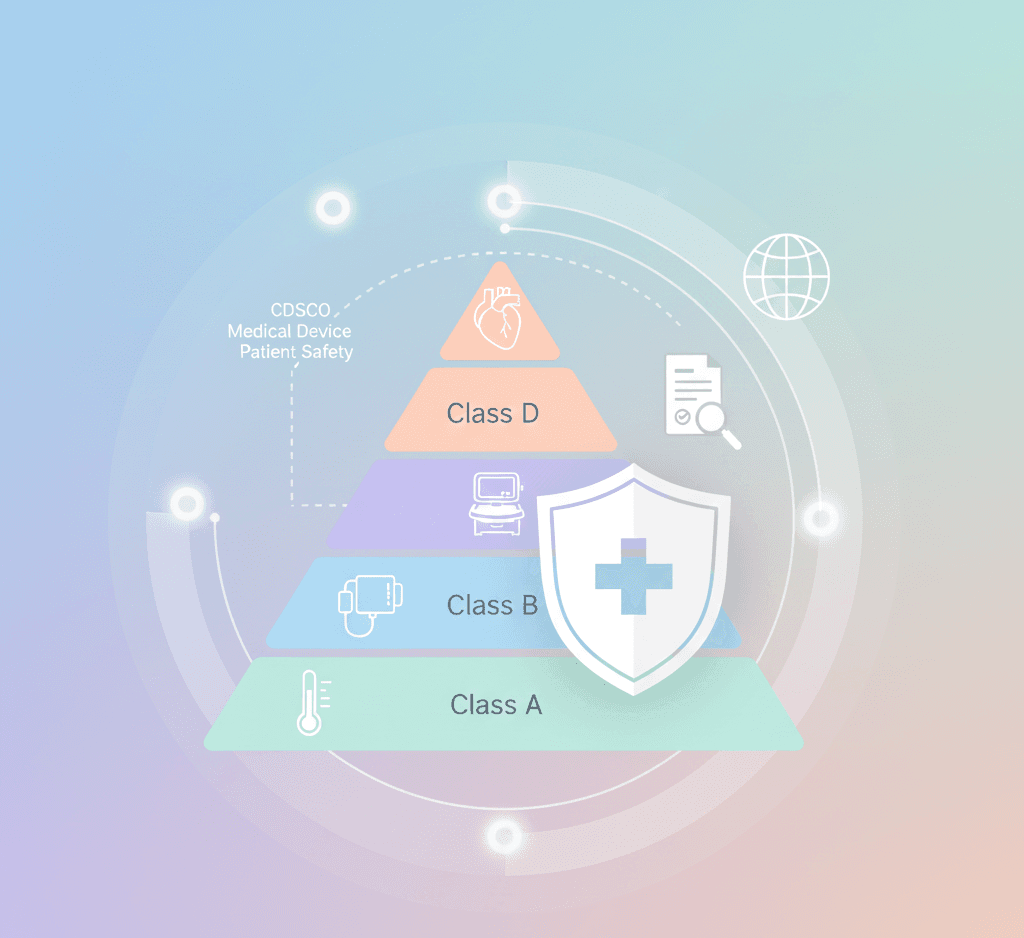
1. Comprehensive Classification System
Devices are now strictly classified into Class A (low risk) to Class D (high risk), with regulatory scrutiny increasing by class. Manufacturers must ensure that device classification aligns with intended use and risk profile before filing applications.
2. Import License & Registration Renewal
The CDSCO has digitized import license renewals, reducing manual delays. However, compliance documentation—especially testing data and Free Sale Certificates—must now align with the latest international standards (ISO 13485, IEC 60601).
3. Mandatory Local Representation
Foreign manufacturers must appoint an Authorized Indian Representative (AIR) to handle regulatory submissions and post-market activities. Non-compliance may lead to license suspension.
4. Audit & Quality System Strengthening
Periodic audits by notified bodies are now stricter. Manufacturers are expected to maintain a full Quality Management System (QMS), with clear SOPs, traceability, and documentation on complaint handling and vigilance.
5. Increased Post-Market Surveillance
Post-approval vigilance reporting has become mandatory. Companies must report adverse events and corrective actions within specified timelines.
Impact on Manufacturers and Importers
The 2025 changes emphasize accountability at every stage—from design to distribution. For manufacturers, this means:
Higher documentation standards: Detailed technical files and clinical data.
Operational adjustments: Continuous monitoring, internal audits, and revalidation.
Longer approval cycles (initially): But faster renewals once compliant.
Need for expert regulatory partners: To manage applications and interface with authorities.
For importers, maintaining updated regulatory documents and liaising with foreign suppliers for ISO compliance has become essential.
How to Prepare for the New Regulatory Era
1. Conduct a Compliance Gap Assessment
Evaluate your current documentation and licensing status against the latest CDSCO and BIS requirements.
2. Strengthen Your QMS
Implement ISO 13485-compliant systems covering design control, production, testing, and complaint handling.
3. Reassess Device Classification
Ensure your product is categorised correctly. Misclassification is one of the top reasons for regulatory rejection.
4. Update Technical Documentation
Align product dossiers, risk management files, and labelling as per MDR 2017 and the latest amendments.
5. Train Regulatory & Quality Teams
Keep staff updated on process changes, import rules, and documentation formats.
6. Partner with Expert Consultants
Work with experienced MedTech advisors who understand the nuances of CDSCO and international compliance frameworks.
Example Scenario: Class C Device Registration
A startup manufacturing a diagnostic imaging device must now register under Class C, requiring:
ISO 13485 certification,
Clinical performance evaluation,
Authorised Indian Representative appointment (if imported),
Application submission via the SUGAM portal,
Post-market vigilance reporting every six months.
This process, while structured, ensures global-level safety and traceability—helping manufacturers expand internationally.
The Bigger Picture: Global Alignment
India’s 2025 regulatory shift isn’t just local—it’s a step toward harmonisation with global standards like US FDA, EU MDR, and WHO benchmarks. This alignment boosts investor confidence and export readiness while protecting patient interests.
Final Thoughts
The evolution of India’s medical device regulations signals a maturing MedTech ecosystem—one that prioritises safety, transparency, and innovation. For manufacturers and importers, the key to success lies in proactive compliance.
By adopting a structured, quality-driven approach today, you not only meet CDSCO’s requirements but also strengthen your brand’s credibility in global markets.
If you’re navigating the new regulatory landscape, our MedTech experts can help assess, prepare, and file your applications efficiently—ensuring your devices stay compliant and market-ready.